Status number - a useful tool for tree breeding
Dag Lindgren1) and Kyu Suk Kang1), 2)
1)
SE-901 83 Umeå, Sweden
2)
Forest Genetics Research Institute, P.O. Box 24, Suwon, Kyonggido, 441-350, Korea
Summary
The concept status effective number (status number) is reviewed. The status number of a given population is the number of unrelated and non inbred trees which shares the following characteristics with the population under study: degree of average relatedness; probability that genes taken at random are identical by descent; expected drift of gene frequencies; gene diversity and expected heterozygosity; expected inbreeding in the progeny following random mating; and often a similar number of rare alleles. It is desirable that tree breeding programs monitor and manage both the genetic gain and status number. Status number is a useful tool for evaluating the consequences of different breeding strategies and for balancing gain versus gene diversity (=relatedness).
Key words: effective number, genetic diversity, gene diversity, genetic gain, coancestry, kinship, relatedness
Introduction
A quantitative measure of gene diversity is required to monitor what happens in the breeding populations and to manage the gene resources considering gene diversity. The actual number of a breeding population does not give sufficient information about the gene diversity in it. So additional measurements are needed. Wright (1931) proposed the effective population size concept as a means of relating observed or expected numbers and population properties. Expected contributions of families or individuals to the gene pool (from which the next generation is drawn) of the progeny population can be used to determine the effective population size (Robertson 1961).
We would like to focus on genetic resources, which may be decreased or lost by forest tree breeding. Tree breeders select a small number of founders, which will be used to a variable but often high extent. Genes from some founders become abundant, while genes from others become rare or sometimes lost. These processes are likely to cause a loss of gene diversity. Gene diversity is the raw material for breeding; this is an additional reason why the possible reduction of genetic diversity is an important concern for tree breeders. Sustainable long-term breeding is dependent on preservation of gene diversity. But genetic drift leads to stochastic changes. Alleles are lost and inbreeding and coancestry increase. The classic effective population size concept handles this situation well for a situation where forces act over a number of generations, but is less suitable for a description what happens in one or a few unique generation shifts. Forest tree breeding is a young science, which is still close to the "wild" forest, and uses a different tactics for different generation in the beginning. Thus status number has been suggested as an alternative effective number, which may meet the needs of forest tree breeders (Lindgren et al. 1996).
The purpose of present study is to describe status effective number and some related concepts, and to review its current and potential use as a breeding tool.
Theoretical background
Neutral genes, diploid zygotes and haploid gametes are assumed in the following. It is useful to understand a number of other concepts for understanding the status number concept . Coancestry (synonymous with coefficient of kinship) is the probability that a gene taken at random from an individual will be identical to a random homologous gene from another individual by descent. The coancestry measures relatedness among two individuals, it becomes the inbreeding of their progeny if they are to mate. The coancestry concept is also applicable if the individuals considered are identical (self-coancestry). We would like to describe how related are the individuals in a group of individuals. A logical step would then be to consider the average of all possible relationships in the group. Such a concept was defined by Cockerham (1967), referred to as group coancestry, while others called it average coancestry or average kinship (e.g. Ballou and Lacy 1995; Lindgren et al. 1996), being the average of all pair-wise coancestries, including self-coancestry and reciprocals. Average coancestry is, however, frequently used for other types of averages (e.g. excluding self-coancestry), and thus we prefer the terminology group coancestry. The group coancestry becomes the expected average inbreeding of the group's progeny following random mating. Thus, group coancestry is associated with inbreeding, but group coancestry is not a measure or prediction of inbreeding. The group coancestry is also the probability that two genes taken at random from the gene pool are identical by descent (Cockerham 1967). It follows that the likelihood that two genes sampled from the gene pool will be identical by descent is half the inverse of the status number. Note that this pairing of gametes for comparing if they are identical by descent is not the same thing as mating or formation of zygotes.
It is important to notice that the concepts of inbreeding, coancestry and group coancestry all make comparison to a reference population, which by definition is without inbreeding or relatedness. Even if it seldom exists, it is an important concept, which is realised by considering that coancestry and inbreeding does not make sense without it. For group coancestry, it is only the gene pool in the population considered that matter, not how those genes are distributed among individuals. Group coancestry can be considered as a characteristic of the gene pool of the group. It is independent of how genes are combined in zygotes or how they will recombine in the progeny, this means that it gives a measure of the potentiality of such a gene pool.
An effective number is more conventional and more understandable than group coancestry; therefore, Lindgren et al. (1995, 1996, and 1997) developed the concept of "status number". Let us first introduce symbols to be used in our discussion, group coancestry will be denoted ![]() . The status number, Ns, is defined as half the inverse of group coancestry, thus
. The status number, Ns, is defined as half the inverse of group coancestry, thus ![]() . Note that the inverse of group coancestry may be viewed as the effective number of founder genes, this may be a useful concept, but will not be discussed further here. The factor 0.5 is introduced, as there are two sets of genes per zygote.
. Note that the inverse of group coancestry may be viewed as the effective number of founder genes, this may be a useful concept, but will not be discussed further here. The factor 0.5 is introduced, as there are two sets of genes per zygote.
Let us consider a gene pool and studying the likelihood that two genes sampled from this gene pool are identical by descent. The gene pool is viewed at as the sum of genes from a number of contributors. The probability that the first gene originates from genotype i is pi, and the probability that the second gene originates from genotype j is pj. The likelihood that these two genes are identical by descent is ![]() , where
, where ![]() is the coancestry between genotype i and j. The probability that a pair of genes sampled from the joint gamete gene pool are identical by descent is found by adding over all possible pairings of gametes from n contributors, thus
is the coancestry between genotype i and j. The probability that a pair of genes sampled from the joint gamete gene pool are identical by descent is found by adding over all possible pairings of gametes from n contributors, thus
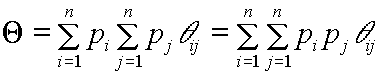 [1]
[1]
Formula [1] illustrates the dual nature of ![]() , the expression appears both with a probabilistic and with an arithmetic (weighted average of coancestry) definition of group coancestry. As coancestry is a probability, it is not too surprising that average coancestry can also be interpreted as a probability.
, the expression appears both with a probabilistic and with an arithmetic (weighted average of coancestry) definition of group coancestry. As coancestry is a probability, it is not too surprising that average coancestry can also be interpreted as a probability.
The contributors to the gene pool could be viewed at as parents and the p: s could be viewed at as the fraction among all successful gametes. In the seed crop from a seed orchard there are many successful gametes, the gene pool is large, so there is no genetic drift, and the contributions to the gene pool can be evaluated from the expected fraction of contributions only.
It is also possible to view "the contributors" as the population under study. For this case individuals contribute equally to the gene pool, p=1/n and group coancestry will be
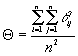 (That is the average of all individual coancestry values in the population).
(That is the average of all individual coancestry values in the population).
If the genotypes have no relatedness and no inbreeding, formula [1] is simplified to be
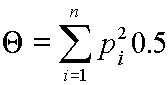
and thus
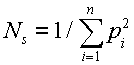 [2]
[2]
This special case is familiar from other work and has often been interpreted as an effective size or diversity related measure (e.g. Robertson 1961; Kimura and Crow 1963; Kang and Namkoong 1988; Wei 1995). There are conventional expressions "effective size of a population" (e.g. Falconer and MacKay 1996). These measures use the concept "population" in a dynamic meaning, while it is used in a static meaning here. They measure how inbreeding accumulates or gene frequencies disperse in a population by time as the members of the population changes, while the status number refers to a specific population. The conventional effective numbers are useful for describing what happens over a number of generations under similar conditions, but are less useful for forest tree breeding, which is still in the first generations and where the strategy changes fast by time.
It is useful to regard the conceptual reference population as infinite in size, as then the status number can be regarded as the size of a sample from the reference population which has the same characteristics as a studied populations from some important prospects. Status number cannot be bigger than the census number and not smaller than 0.5 (the value for a gamete).
An example of group coancestry and status number is given, it helps the reader to develop a feel for the concept if this simple example is solved. A pair of full-sibs; or a mother and her child; or two unrelated trees originating from selfing, all have ![]() .
.
Group coancestry forms a logical addition to a hierarchy of concepts, which can be formulated:
Although status number and group coancestry are informative concerning the genetic status of a population, they do not yield complete information about the value of a population as a gene resource. If original genomes are unequally distributed in the population, status number can theoretically raise at a generation shift. Thus the maximum status number derivable from a population is of interest, but this concept has not been well developed yet. We can not express a mathematical relation between status number and rare alleles, although still status number is rather informative on rare alleles.
Status number and gene diversity
Gene diversity, also termed expected heterozygosity, is a common and useful measure of genetic variation within a population (Wright 1969; Nei 1973, Lacy 1995). Gene diversity is the variance in allele frequencies at a genetic locus and is equal to the heterozygosity expected in a population with random union of gametes (in Hardy-Weinberg equilibrium). For a single genetic locus, gene diversity can be formulated as
![]() [3]
[3]
in which qi is the frequency of allele i and the summation is over all alleles at that locus (Lacy 1995, Weir 1990). Gene diversity can be averaged over loci to provide a genome-wide metric. We consider here the case that the diversities are related to a population of founders where each individual has two unique alleles.
Status number in tree breeding
Askew and Burrows (1983) derived the formula for status number but they thought it was variance effective number. They made numerical calculations concerned with the effect of selection in progenies of Pinus taeda and it could be converted to status number. For seed orchards (comprising what can be considered as a sample from the reference population) with non related and non inbred clones, the status number is sometimes equivalent with earlier formulations like effective number of clones or diversity (e.g. Fries 1994
; Lindgren and El-Kassaby 1989).Mullin and Park (1995) made a stochastic forest tree breeding simulator (POPSIM) with status number calculations as an option. This simulator makes it possible for tree breeders to study the consequences of different tree breeding strategies in terms of gain and status number. It is also possible to compare the concept of status number with the effective population size in the inbreeding sense for the same program. Lindgren et al. (1995,1996,1997) suggested that the concept of status number would be a useful measure to monitor diversity in tree breeding. Simple situations can be evaluated with formulas, like the status number as a function of family size and type; and how status number develops over time in a small closed breeding system (Lindgren et al. 1996); or the decay of status number over generations in simulated or real breeding programs (Lindgren et al. 1997).
The basic outcome from a tree breeding strategy is gain and gene diversity. Gene diversity can be expressed in terms of relatedness or status number according to formula [3]. Thus it is functional to plot gain and status number against each other and compare gain at the same status number. This technique has been used to compare different tree selection and breeding strategies using Monte-Carlo simulations (Spanos et al. 1997; Andersson et al. 1998 a and b). The results indicate that phenotypic selection is efficient when selecting among the offspring. If relatives are considered when predicting breeding values, and a combined index between the family and the individual is used for selection, the gain resulting from a selection can be increased, but this is accompanied by a decrease in status number. The gene diversity can be improved for combined index selection if restrictions for the highest number of contributions from families or parents are made, but if the diversity is increased in that way, the gain sinks correspondingly. If compared at the same selected proportion and status number, phenotypic selection is better and at least not significant inferior to combined index selection combined with a restriction. Phenotypic selection is less favourable at low heritability and large families, however. The results also demonstrate that strategies which maximise gain leads to unacceptable low status number, thus tree breeding without considering status number (or another formulation of diversity) is unlikely to be sustainable, but will soon lead to unacceptable levels of relatedness. Wei et al. (1997) developed predicative formulas for gain and status number in a situation with restricted phenotypic selection or restricted combined index selection. The setting was a single round of selection in a population comprising unrelated families without inbreeding. The restrictions could apply to as well the number of families selected as the number of individuals within any family. In the given total population size, a proper balance between family number and size was important to get a high gain at a reasonable status number. Selecting from the better families only boosted the gain from phenotypic selection and restricting the number of selections per family boosted the status number for combined index selection. Theoretical prediction gave results close to what was observed when the selection algorithms were applied to real materials.
Gain and status number can be studied separately. However, an optimal breeding or selection strategy can be devised if the entities are compatible on the same value scale (thus the value of a certain change in gene diversity can be regarded as equivalent to that of a change in gains, e.g. volume production). As gene diversity is equivalent to relatedness and as relatedness is the cause of inbreeding, there is a rationale for such a common value scale. Lindgren and Mullin (1997) have shown the superiority of such a "population merit" selection using Monte-Carlo simulations, where both gain and status number were considered, over a more conventional approach to limit the loss of gene diversity by constraining parental impact. The superiority was considerable and growing in later generations of a multiple generation breeding program.
Mullin (1997) applied the algorithm to optimise selection of a second-generation breeding population of jack pine in Ontario, Canada. A population of 350 trees was to be chosen from over 32 000 OP test progeny. Population-merit solutions always gave superior gain at a given status effective number, compared to various approaches to restricting family contributions, and produced 5.4% more gain than a Poisson distribution of family contributions suggested in the current breeding strategy. Zheng et al. (1997) reported similar analyses with real data concerned with clone selection of Swedish Norway spruce suggesting a selection algorithm, which maximised population merit. Explicit formulations of a selection criteria for individuals considering coancestry were given. The optimal number of selections was dependent on the relative value of status number versus gain. Both studies used a trial and error method to find the optimum and the superiority was robust against different assumptions on the relative value gain and status number (gene diversity).A measure of diversity (or effective number) is desired for the crop from seed orchards. Lindgren and Mullin (1998) applied status number to seed orchard crops as a function of the inbreeding, relatedness, male fertility and female fertility of seed orchard clones. The applications were extended to the influence of pollen contamination as a function of the percentage of contamination and the relatedness among the contaminating pollen sources as well as between pollen source and orchard clone. Kjaer and Wellendorf (1998) compared status number to other measures of effective population size for a Danish Norway spruce clonal seed orchard and its crop. The census number of seed orchard clones was 100. The status number was 70.4; the variance effective population size 236.7 and the inbreeding effective population size 18.1, respectively. Kang and Lindgren (1998) registered the amount of male and female flowering among clones in two seed orchards and one clonal archive in Korea (these are considered as seed orchard populations and are identified by the species). Assuming the observed clonal differences in flowering were representative for the fertility of seed orchard clones, the seed orchard crops could be predicted as follows:
Table 1. Group coancestry, status number, relative status number, and relative gene diversity of the predicted
orchard crops based on the fertility variation in three considered clonal seed orchard populations.
|
Seed orchard |
Number of clones |
Group coancestry |
Status number |
Relative status number |
Relative gene diversity |
|
Pinus densiflora |
99 |
0.00722 |
69.3 |
0.70 |
0.993 |
|
P. thunbergii |
60 |
0.00915 |
54.6 |
0.91 |
0.991 |
|
P. koraiensis |
180 |
0.01289 |
38.8 |
0.22 |
0.987 |
The predictions of status number in the future crops are similar to some earlier concepts of "effective number of clones in a seed orchard". It was noted that the number of male flowers was not correlated with the number of female flowers in three objects and lower status number would probably apply in a seed orchard with a stronger correlation. The last object is rather young; it is likely that status number of the crop is lower in a young seed orchard than a mature. The decrease in gene diversity compared to the reference population (that can be seen as the wild forest from which the plus trees were collected and has the value 1, that is why we call it relative gene diversity) does not look alarming.
Sometimes earlier concepts "effective population size" or "diversity" are equivalent to recently defined status number when applied to the problem of optimising first generation seed orchards (Lindgren and Matheson 1986; Bondesson and Lindgren 1993) or clonal mixtures (Lindgren et al. 1989). The number of ramets should be a linear function of the breeding value of the clone to get maximal gain at a given status number. This is valid for the special case that clones are unrelated and not inbred. These results may also serve as demonstration of the results achievable by the technique to maximise gain at a certain status number.
A breeding program often needs fresh material. A model for studying the effect of infusion of fresh material into the breeding population was developed by Zheng et al. (1998). The model assumes where is a large pool of untested, unrelated and non-inbred plus trees, and a bred material comprising full sibs from a number of superior plus trees. The subject of study is to what degree it increases the population merit to choose unrelated but unimproved plus trees instead of selections among the offspring that generally is genetically better but more related. It seemed profitable to include some percent of new plus trees rather than selecting among the progeny of all plus trees. The percentage is higher the lower the effect of plus tree selection (and a subsequent progeny testing using other individuals than that selected). The status number of modern apple cultivars is 8, this indicates clearly that apple tree breeders are operating with a population of greatly reduced diversity (Noiton and Alspach 1996).
Via the home page http://linne.genfys.slu.se/breed/breed.htm an FTP site managed by Dag Lindgren can be reached, where programs helpful in understanding and applying techniques described in this review are made available.
Founder genome equivalents
Lacy (cf. Lacy 1995) defined the concept "founder genome equivalent" for applications in captive populations (mainly animals in zoological gardens) with the objective to preserve genetic diversity. The first generation of captives which reproduce in captivity are seen as founders, and the founders are seen as a sample from a source population, which is an analogue to the reference population. Founder genome equivalent has two non-identical definitions (Lacy 1995, p568; Ballou and Lacy 1995), the first one will be called variety "remaining allele related" and the second will be called variety "status number analogue". The second formulation was preferred by Lacy (1995), who explained it as "pedigree analysis of mean kinship". The first concept was defined as an effective number of the remaining founder alleles in the population under study. The second concept was derived as the number of founders, which harbour the same gene diversity as the population under study. The later one is more or less equivalent to status number if the reference population is equivalent to the source population from which the founders are drawn. The first concept is a rather good approximation for the second, the approximation seems to work worse the smaller and simpler the population is (Lacy 1995). For two full sibs status number is 1.33 but "founder genome equivalents var. remaining allele related" is 1.5. The first concept is not practically possible to calculate accurately for a complicated pedigree, instead it must be estimated by a Monte-Carlo simulation method. Founder genome equivalents are based on a somewhat specific and restricted definition of founders and the source population from which the founders can be regarded as a sample. The problem with possible relatedness and inbreeding in the source population is removed by definition; inbreeding, coancestry, founder allele loss and genetic diversity are related to those in the source population. The status number formulation is more suitable for a study of some of these important phenomena (as done by Lindgren and Mullin 1998). No hit on founder genome equivalents (neither founder genome) was found in a search for the last year+ in Biological Abstracts, even those who cite and use the concept sometimes do it under other names. The meaning of genome in "Founder genome equivalents" could easily be misinterpreted (as founder gene equivalents as mentioned above) as in some genetic texts a diploid has two genomes and in some one.
Boichard et al. (1997) calculated "founder genome equivalents var. remaining allele related" for three French cattle populations (the figures were based on in total 3,364,386 pedigreed animals) to be 17 in Abondance, 22 in Normandie and 206 in Limousine, respectively. Safari and James (1998) carried out a similar work using the same measure as above for Merino sheep selected populations. The captive populations in zoological gardens were characterised by the founder genome equivalent which was the variety of status number analogue to 6.0 for Okapi and 10.0 for Goeldi's monkey (Lacy 1995). These measures of captive populations had the theoretical potential to be raised to 11.4 and 16.0, respectively, by mating so a more equal distribution of founder genes in the population would be achieved. Sánchez (1997) used "founder genome equivalents var. remaining allele related" as a method to compare the maintenance of gene diversity between random mating and mating selection lines in Drosophila melanogaster. It was seen after 6 generations of intense selection a considerable reduction in that measure, from 120 initial founders to 4.5 as average at the end of the experiment, and a slight difference between lines.
Ways to visualise status number
Status number is a convenient way of expressing group coancestry in terms of an effective population size. Status number of a population can be seen as the number of genotypes sampled from a reference population (a large population without inbreeding and coancestry) which share the following characteristics with the studied population:
Status number expresses the accumulated genetic drift from the same reference population to which the concepts inbreeding and coancestry refers.
Conclusion
Status number is a useful tool for monitoring and managing the populations dealt with by tree breeders. Tree breeders are also interested in gain, by joint consideration of gain and status number they can both utilise and conserve the genes.
Acknowledgement
The development and implementation of the status number concept has been a joint work over some years and appreciated input was made by many persons, among others: Bengt Andersson, Erik W Andersson, Adolfo Bila, Lennart Bondesson, Rowland Burdon, Ola Rosvall, Tore Ericsson, Öje Danell, Luigi D Gea, Paul Jefferson, Erik D Kjaer, Bill (WJ) Libby, Tim (TJ) Mullin, Gene Namkoong, Run-Peng Wei, Leopoldo Sánchez, Kostas Spanos, Tony (ACT) Shelbourne, Yong-Qi Zheng,
Literature Cited
Lindgren, D., Wei, R.-P. and Lee, S. 1997. How to calculate optimum family number when starting a breeding program. For. Sci. 43(2): 206-212.
Mullin, T.J. 1997. Lake Nipigon West Breeding Zone jack pine family tests: selection of the second-generation breeding population. Contract report for the Ontario Tree Improvement Board, Sault Ste. Marie, ON. 8 pp. (+ appendices).